Theodora Markati, Jessica Duis & Laurent Servais
Páginas 709-720 | Recibido el 19 de marzo de 2021, aceptado el 3 de junio de 2021, aceptado la versión del autor publicada en línea: 10 de junio de 2021, publicada en línea: 28 de junio de 2021
RESUMEN
Introducción: El síndrome de Angelman es un trastorno genético del neurodesarrollo poco común, causado por la deficiencia o función anormal de la ubiquitina proteína ligasa E3A materna, conocida como UBE3A, en el sistema nervioso central. No está disponible ningún tratamiento que modifique la enfermedad, pero la línea terapéutica del síndrome de Angelman incluye al menos 15 enfoques diferentes en el desarrollo clínico o preclínico. En los próximos años, varios ensayos clínicos incluirán pacientes, lo que motivó esta revisión integral.
Áreas cubiertas: Resumimos y revisamos críticamente los diferentes enfoques terapéuticos. Algunos enfoques intentan restaurar la proteína UBE3A inexistente o no funcional en las neuronas mediante terapias de reemplazo de genes o de reemplazo de enzimas. Otras terapias tienen como objetivo inducir la expresión de la copia paterna normal del gen UBE3A apuntando con un ARN largo no codificante, el UBE3A-ATS, que interfiere con su propia expresión. Otra categoría terapéutica incluye compuestos que se dirigen a rutas moleculares y proteínas efectoras que se sabe que están involucradas en la patofisiología del síndrome de Angelman.
Opinión de expertos: Creemos que para 2022–2023, más de cinco tratamientos modificadores de la enfermedad estarán simultáneamente en pruebas clínicas. Sin embargo, existen varios desafíos con respecto a la seguridad y la eficacia, que deben ser abordados. Además, todavía existe una importante necesidad insatisfecha de preparación para ensayos clínicos.
PALABRAS CLAVE
Virus adenoasociado, síndrome de Angelman, oligonucleótido antisentido, terapia celular, CRISPR-CAS9, terapia génica, impronta genómica, UBE3A, UBE3A-ATS, dedos de zinc.
- Introducción
El síndrome de Angelman (SA), caracterizado por primera vez por el Dr. Harry Angelman en 1965, es un trastorno genético del neurodesarrollo poco común diagnosticado en uno de cada 12.000–20.000 nacidos vivos (NORD, 2018 y OMIM 105830). Los pacientes con SA presentan un retraso global del desarrollo, dificultades de aprendizaje y un retraso particularmente severo del lenguaje expresivo. En cuanto al comportamiento, los pacientes tienen un comportamiento característicamente feliz, que generalmente se expresa como una risa no provocada, devoción por el agua y un comportamiento desadaptativo. Los pacientes tienen trastornos del movimiento, que incluyen ataxia de la marcha, temblor de las extremidades e hipotonía generalizada del tronco. Por lo general, los pacientes también presentan actividad convulsiva y alteraciones del sueño [1–8]. El tratamiento es de apoyo y se centra en las convulsiones, el sueño y el comportamiento, ya que actualmente no hay disponibles tratamientos modificadores de la enfermedad o específicos para el SA.
La causa del SA es la deficiencia o función anormal de la ubiquitina-proteína ligasa E3A, conocida como UBE3A, que se expresa a partir del alelo materno UBE3A, ubicado en el cromosoma 15 en humanos [9,10]. La pérdida de expresión del UBE3A materno se produce a través de varios mecanismos moleculares. Con mayor frecuencia, se produce por la deleción de novo del área crítica materna 15q11.2-q13 en el cromosoma 15 (aproximadamente el 75% de los casos) [11]. Otras causas incluyen cambios de marco, mutaciones sin sentido o sin sentido en UBE3A, disomía uniparental paterna y defectos de impronta [5,10]. El UBE3A está impreso en el sistema nervioso central (SNC), en el que la copia paterna es silenciada por una larga transcripción antisentido no codificante, la UBE3A-ATS. Tanto en humanos como en ratones, la transcripción antisentido silencia la producción del gen paterno UBE3A [12, 13].
UBE3A cataliza la ubiquitinación, un proceso mediante el cual las proteínas se etiquetan para su degradación en el proteosoma [14, 15]. Se han identificado varios sustratos candidatos de UBE3A, incluido el calcio (Ca2+) activado por una pequeña conductancia del canal de potasio SK2, efexina-5, p53 y p27 [16,17]. El análisis de redes de UBE3A sugiere que varias vías moleculares podrían contribuir potencialmente a la fisiopatología del SA [18]. UBE3A juega un papel fundamental en la plasticidad sináptica dependiente de la actividad durante el desarrollo [19]. Los modelos de ratón de SA presentan anomalías morfológicas en la columna dendrítica, alteración de la potenciación a largo plazo (LTP) [20-22] y un fenotipo atáxico [23].
Se están siguiendo tres estrategias en el desarrollo clínico y preclínico para el tratamiento del SA. Una estrategia tiene como objetivo restaurar la proteína UBE3A faltante o no funcional en las neuronas mediante terapias de reemplazo de genes o de reemplazo de enzimas. El objetivo de un segundo enfoque es «de-silenciar» la copia paterna del gen UBE3A. El tercer enfoque involucra compuestos que se dirigen a rutas moleculares y proteínas efectoras que se sabe que están involucradas en la patofisiología del SA. La amplia gama de enfoques mecanicistas y el rápido ritmo de descubrimiento hacen que la comprensión de la línea básica actual sea un desafío. Un problema adicional para los médicos en contacto con las familias es que hay datos en dominios públicos, como redes sociales o sitios web de grupos de defensa de pacientes, que no se han publicado en la literatura médica revisada por pares. Esto puede dificultar la prestación de un asesoramiento sólido y actual a los pacientes y la gestión de sus expectativas. El objetivo de esta revisión es resumir los tratamientos y terapias candidatos en las etapas de clínica y etapas finales de preclínica mediante la descripción no solo de las publicaciones revisadas por pares, sino también de todos los datos disponibles públicamente.
- Materiales y métodos
Realizamos una revisión exhaustiva de las publicaciones en PubMed y Cochrane utilizando las palabras clave «Síndrome de Angelman» o «Angelman» y «terapia / -as», «tratamiento / -s» o «terapéutica / -s». También se realizaron búsquedas en todos los ensayos clínicos y estudios en curso registrados en ClinicalTrials.gov mediante el uso de la palabra clave «Síndrome de Angelman». Toda la información disponible públicamente de la cumbre anual de la Fundación para la Terapia del Síndrome de Angelman y la GALA (mencionada en el texto como Cumbre FAST) disponible en el enlace:
https://www.youtube.com/channel/UCuAoKMiWQXb_OcBnZppzQrQ fue usado.
Además, revisamos la información disponible públicamente de los sitios web oficiales de la Fundación para la Terapia del Síndrome de Angelman (FAST) y la Fundación del Síndrome de Angelman (ASF).
[Como consecuencia, los datos presentados a continuación se recopilan no solo de publicaciones revisadas por pares, sino también de comunicados de prensa o presentaciones públicas dadas en varias conferencias por los investigadores principales o representantes de la industria. Las fuentes no revisadas por pares se enumeran como «fuentes adicionales» para indicar claramente que no han sido revisadas por pares.]
- Resultados
3.1. Terapias de reemplazo de genes / enzimas
Varios fármacos en desarrollo para el SA tienen como objetivo restaurar la proteína UBE3A que falta o que no es funcional en las neuronas mediante el reemplazo de genes o el reemplazo de enzimas. Los compuestos, las empresas o las instituciones involucradas, así como las etapas de desarrollo se enumeran en la Tabla 1.
Tabla 1. Terapias de reemplazo de genes / proteínas

- Reemplazo de genes mediado por virus adenoasociado
Daily et al. [24] proporcionaron la primera prueba de concepto de que el SA se puede tratar suministrando exógenamente una copia del gen UBE3A que codifica la proteína homónima a las neuronas. En los experimentos que se conocen, ratones carentes del gen materno Ube3a debido a una mutación nula recibieron inyecciones hipocampales directas de un virus adenoasociado (AAV) serotipo 9, AAV-9, que se había transformado para llevar una copia del gen Ube3a murino [21,24]. Los ratones que recibieron terapia de reemplazo génico mostraron una mejora significativa en el aprendizaje asociativo y la memoria en comparación con los ratones de control, que fueron inyectados con un vector AAV-9 que llevaba el transgén que codifica una proteína verde fluorescente. Sin embargo, a diferencia de la memoria, la LTP no fue rescatada por completo, como reveló la electrofisiología. Además, no hubo una transducción adecuada del transgén en el cerebelo y, por lo tanto, los déficits motores, que se cree que están asociados con esta parte del cerebro, no mejoraron [24,25].
En principio, los vectores AAV, transformados para llevar una copia del gen que requiere reemplazo, son reconocidos por los receptores de la superficie celular de las células diana y se internalizan mediante endocitosis. Luego se trafican intracelularmente en vesículas endosomales y, después de ingresar al núcleo a través del complejo de poro nuclear, su genoma se libera (desencadenamiento). El ADN monocatenario se somete a una síntesis de la segunda cadena utilizando la polimerasa del huésped, ya que se requiere una cadena doble para la transcripción; algunos AAV son «autocomplementarios». Después de esto, el genoma generalmente se estabiliza como episomas circulares, que luego se pueden transcribir a ARNm y traducir a proteínas por la maquinaria celular. El genoma de AAV también puede integrarse en el huésped a baja frecuencia (Figura 1) [26, 27]. Los investigadores se están centrando en la ingeniería de vectores AAV para enfermedades del SNC con mejor potencial de biodisponibilidad y capacidad de transducción neuronal. Además, muchos factores pueden afectar la eficacia de una terapia de reemplazo génico para SA, incluido el uso de diferentes promotores, áreas reguladoras o transgenes UBE3A. Más específicamente, UBE3A codifica las tres isoformas de UBE3A, que pueden ocurrir mediante empalmes alternativos; aún se desconoce si algunas isoformas son más críticas en la fisiopatología del SA [28]. Sobre la base de las presentaciones de la Cumbre FAST anual (fuentes adicionales 1–5), varias instituciones y empresas están trabajando para lograr una terapia de reemplazo génica eficaz para el SA. Un enfoque de terapia génica, que utiliza un vector AAV modificado (USF-AAV), GT-AS (anteriormente conocido como AGIL-AS), está en fase final del desarrollo preclínico.
Figura 1. Terapia génica in vivo y terapia genética ex vivo (terapia celular). En la terapia génica mediada por AAV, los vectores AAV, transformados para llevar una copia del gen que requieren reemplazo, son reconocidos por los receptores de la superficie celular de las células diana y se internalizan a través de la endocitosis. Luego se trata de tráfico intracelularmente en vesículas endosomales y, después de ingresar al núcleo a través del complejo de poros nucleares, su genoma se libera (sin recubrir). El ADN simple se somete a la segunda síntesis de la cadena utilizando la polimerasa huésped, como se requiere una doble cadena para la transcripción; Algunos AAV son «auto-complementarios». Siguiendo esto, el genoma generalmente se está estabilizado como episomas circulares, que luego se pueden transcribir al ARNm y traducir a la proteína por la maquinaria celular. El genoma AAV también puede integrarse en el huésped a baja frecuencia. En la terapia celular, los HSC están aislados del paciente. La programación Ex vivo lentiviral de estos HSC lleva a la integración del gen UBE3A en el genoma. Después del trasplante autólogo, los HSC que llevan la copia normal del gen se diferencian en celdas que tienen la capacidad de cruzar la barrera hematoencefálica. Una vez, en exceso en proceso en el SNC, las células producen proteínas UBE3A y suministran las neuronas deficientes a través de la corrección cruzada.
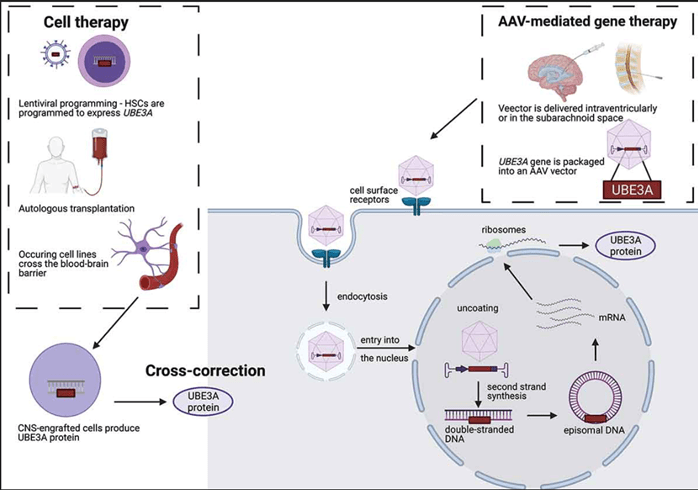
3.1.2. Terapia celular
Una terapia génica, basada en el trasplante autólogo de células madre hematopoyéticas (HSC) después de la inserción ex vivo mediada por lentivirales del gen UBE3A, se encuentra actualmente en una fase final de desarrollo preclínico (Figura 1) [29]. Este tipo de enfoque se está investigando para varios trastornos genéticos, incluidos los trastornos por inmunodeficiencia [30] y otras enfermedades neurogenéticas como la leucodistrofia metacromática [31].
Según las presentaciones en la cumbre de FAST (fuentes adicionales 6–7), este enfoque inicialmente requerirá la recolección de células madre de sangre periférica de los pacientes. Ex vivo, se insertará una copia normal del gen UBE3A en el genoma de las HSC utilizando un enfoque mediado por lentivirales, y luego las células se volverán a infundir por vía intravenosa en el paciente. Este enfoque requiere quimioterapia para permitir una ocupación adecuada de la médula ósea de las células madre hematopoyéticas programadas. Después de un trasplante autólogo exitoso, las HSC se diferenciarán en líneas celulares que ocurren fisiológicamente, incluidas las células inmunes, que pueden cruzar la barrera hematoencefálica. En el SNC, la proteína UBE3A será secretada por las células injertadas con éxito y será recibida por las neuronas deficientes en un proceso denominado corrección cruzada. Este tipo de terapia tiene dos ventajas principales. En primer lugar, el trasplante autólogo de células editadas ex vivo aumenta las posibilidades de éxito, en comparación con el trasplante alogénico contra el que es más probable una reacción inmunitaria. En segundo lugar, la estrategia probablemente proporcionará un tratamiento permanente, ya que, en teoría, UBE3A se integrará en el genoma de las HSC y, por lo tanto, seguirá estando presente después de las divisiones celulares.
3.1.3. Terapias de reemplazo de enzimas
Las terapias de reemplazo enzimático (ERT por sus siglas en inglés) se utilizan ampliamente para el tratamiento de enfermedades metabólicas asociadas con una deficiencia de una sola enzima o una función anormal, como la enfermedad de Gaucher y la enfermedad de Pompe [32,33]. Un ERT se encuentra actualmente en desarrollo preclínico para el SA. ERT tiene como objetivo entregar una forma purificada de la proteína UBE3A faltante o no funcional en las neuronas, tanto en el espacio intracelular como extracelular. En un estudio reciente en animales, los investigadores encontraron que el UBE3A no solo se excreta, sino que mantiene la actividad enzimática ubiquitinante fuera de las neuronas [34]. ERT todavía está en el nivel de descubrimiento. Se están realizando estudios basados en células y animales para probar el concepto y evaluar la seguridad de dicha terapia (fuente adicional 8).
3.2. «De-silenciar» el alelo paterno
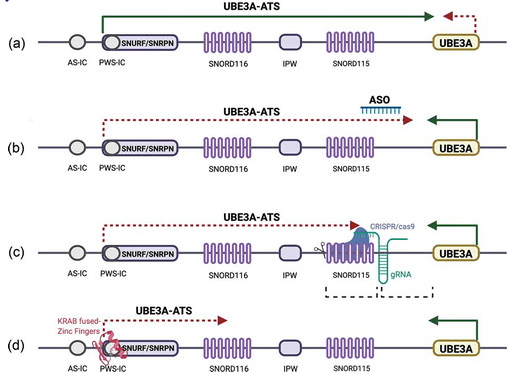
Tanto en humanos como en modelos de ratón, la transcripción de la larga transcripción de ARN no codificante, UBE3A-ATS y Ube3a-ATS respectivamente, está regulada desde áreas en el centro de impresión del síndrome de Prader-Willi o aguas arriba del centro de impronta del síndrome de Prader-Willi (PWS-IC). El Ube3a-ATS se ejecuta a través de Snurf / Snrpn, Snord116, Ipw, Snord115 y hasta la región de codificación de Ube3a en orientación antisentido [35]. En el cromosoma 15 paterno, la transcripción del UBE3A-ATS da como resultado el «silenciamiento» del gen UBE3A (Figura 2a) [12].
Figura 2. Estrategias terapéuticas para “de-silenciar” el UBE3A paterno. (a) La copia paterna normal del gen UBE3A en el cromosoma 15 está «silenciada» debido a la impronta genómica. Tanto en humanos como en modelos de ratón, la transcripción del ARN largo no codificante, UBE3A-ATS y Ube3a-ATS respectivamente, está regulada desde áreas en el PWS-IC o aguas arriba. El Ube3a-ATS pasa por Snurf / Snrpn, Snord116, Ipw, Snord115 y hasta la región de codificación de Ube3a en orientación antisentido. En el cromosoma 15 paterno, la transcripción del UBE3A-ATS da como resultado el «silenciamiento» del gen UBE3A. (b) Los ASO complementarios a la parte distal de UBE3A-ATS pueden conducir a la escisión mediada por RNasa H del híbrido ASO/ARN y a la terminación prematura de la transcripción de UBE3A-ATS. En ausencia de transcripción de UBE3A-ATS, se expresa el UBE3A paterno. Los ASO se encuentran actualmente en desarrollo clínico. (c) La mutagénesis mediada por CRISPR/Cas9 en las áreas genómicas que codifican el UBE3A-ATS puede potencialmente conducir a la «anulación del silenciamiento» del UBE3A paterno probablemente por el cese temprano de la transcripción UBE3A-ATS. Los corchetes indican las áreas que conducen a «de-silenciar» el UBE3A paterno cuando se apunta en estudios con animales. Este enfoque aún se encuentra en desarrollo preclínico. (d) Las proteínas con dedos de zinc fusionado KRAB que se unen al promotor de UBE3A-ATS pueden potencialmente suprimir la transcripción de UBE3A-ATS y «de-silenciar» el UBE3A paterno. Este enfoque aún está en desarrollo preclínico.
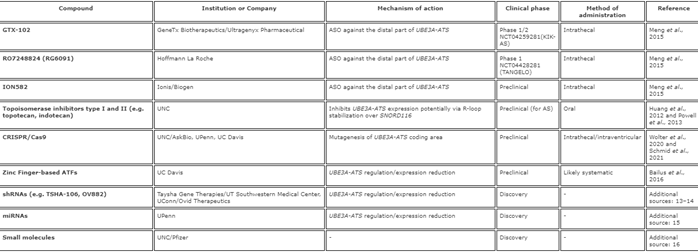
Los niveles de ATS, debidos a la deleción de su área promotora, conducen a un aumento de la expresión de Ube3a paterno [12,36]. Los ratones con un casete de poli (A) entre las áreas Snord115 y Ube3a en el cromosoma paterno, lo que da como resultado la terminación prematura de la transcripción Ube3a-ATS, tienen niveles reducidos de Ube3a-ATS y el doble de ARNm de Ube3a, en comparación con los ratones de control. . Los ratones con carencia de Ube3a materno debido a una mutación nula [21], que recibieron el casete poli (A) en el lado paterno, habían aumentado los niveles de Ube3a en diferentes regiones del cerebro. Estos ratones exhibieron características fenotípicas mejoradas, incluida una coordinación motora mejorada y una mejora de la LTP [13]. Esta fue la primera prueba de concepto de que la inhibición selectiva de la transcripción de UBE3A-ATS puede conducir a «de-silenciar» el alelo UBE3A paterno y desencadenó varios enfoques terapéuticos. Los compuestos, empresas o instituciones involucradas y etapas de desarrollo se enumeran en la Tabla 2.
Tabla 2. De-silenciar la copia paterna
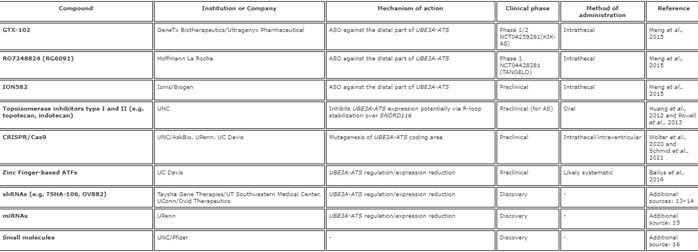
Abreviaturas: ASO: oligonucleótido antisentido, UNC: Universidad de Carolina del Norte, Chapel Hill, AskBio: Asklepios Biopharmaceutical, UPenn: Universidad de Pennsylvania, UC Davis: Universidad de California Davis, ATF: factores de transcripción artificiales, shRNA: ARN corto de la horquilla, UT Southwestern Centro médico: University of Texas Southwestern Medical Center, UConn: Universidad de Connecticut, miARN: microARN
3.2.1. Oligonucleótidos antisentido
El uso de oligonucleótidos antisentido (ASO) complementarios a la parte distal del Ube3a-ATS aumenta la expresión paterna de Ube3a, probablemente mediante el reclutamiento de RNasa H, que degrada el híbrido ASO/RNA. Ube3a-ATS y el crítico para el síndrome de Prader-Willi (PWS), Snord116, se procesan a partir del mismo ARN precursor. Curiosamente, la producción de Snord116 maduro no se ve afectada, probablemente debido a la rápida tasa de empalme en comparación con el tiempo requerido para la transcripción entre el Snord116 y el sitio de unión de ASO (Figura 2b) [37].
Actualmente, dos ASO se encuentran activamente en ensayos en humanos, llamados GTX-102 y RO7248824. Un tercero, ION582, está en desarrollo preclínico. Los ASO pueden diferir tanto en sus secuencias como en sus estructuras [38]. Por ejemplo, los ácidos nucleicos bloqueados (LNA) son un tipo específico de ASO con una estructura no natural que da como resultado una mayor afinidad, una mayor estabilidad metabólica y una menor toxicidad. Los LNA tienen un puente birradicular entre los carbonos C2 y C4 de la ribosa [39].
El primer ensayo clínico de fase 1/2 (KIK-AS, NCT04259281) del ASO, GTX-102 mostró resultados prometedores. Según el comunicado de prensa (fuente adicional 9), cinco pacientes han sido tratados con GTX-102. El fármaco se administró por vía intratecal con un esquema ascendente de cinco dosis. Los participantes presentaron una mejoría clínica que duró al menos de tres a cinco meses. Todos los pacientes mostraron una mejoría al menos en tres dominios de la Escala de Impresiones Clínicas Globales (CGI) ajustadas para el SA. Después del tratamiento, los pacientes habían mejorado las puntuaciones en los dominios de la comunicación receptiva y expresiva en las Escalas de Bayley de Desarrollo de Bebés y Niños Pequeños-4 (BSID-4) y tres de ellas en la herramienta de comunicación Observer Reporter Communication Ability (ORCA). Sin embargo, los cinco participantes presentaron el evento adverso grave de debilidad de las extremidades inferiores a las dosis más altas probadas, que se asoció con la inflamación de las meninges y las raíces nerviosas en la región de la administración intratecal. La debilidad de las extremidades inferiores se resolvió para todos los participantes y los beneficios clínicos observados del tratamiento duraron mucho más que la duración del evento adverso, aproximadamente de tres a cinco meses después de la última dosis.
RO7248824 (o RG6091) se encuentra actualmente en ensayo clínico de fase 1 (TANGELO, NCT04428281) (fuente adicional 10). ION582 está en desarrollo preclínico (fuente adicional 11).
3.2.2. Inhibidores de la topoisomerasa
Durante un proceso de selección de moléculas pequeñas con el potencial de «de-silenciar» el alelo UBE3A paterno, 16 inhibidores de la topoisomerasa tipo I y II mostraron resultados prometedores. El inhibidor de la topoisomerasa I, topotecan, que está aprobado por la Administración de Drogas y Alimentos de los Estados Unidos (FDA) para su uso como agente quimioterapéutico, mostró resultados prometedores tanto in vitro como in vivo [40]. Para el cribado se utilizaron ratones portadores de un gen de fusión entre el Ube3a paterno y un gen codificador de una proteína amarilla fluorescente [20]. In vitro, la administración de topotecan resultó en la restauración de Ube3a funcional a niveles de tipo salvaje en cultivos de neuronas corticales primarias de ratones deficientes en Ube3a materna debido a una mutación nula [21]. Cuando estos ratones fueron tratados con topotecán por vía intracerebroventricular, los niveles de Ube3a aumentaron en el hipocampo, el cuerpo estriado, la corteza cerebral y el cerebelo de una manera dependiente de la dosis [40]. Cuando se administró por vía intratecal, el topotecán aumentó la expresión paterna de Ube3a principalmente en las neuronas de la médula espinal y los resultados se mantuvieron 12 semanas después de la última dosis [40]. Mecánicamente, el topotecán suprime la transcripción de Ube3a-ATS mediante la estabilización de los bucles R sobre el grupo paterno de Snord116, que se sabe que crean inestabilidad genómica y cese de la transcripción [41,42]. Dado que la biodisponibilidad del topotecán en el SNC es limitada, se investigaron otros inhibidores de la topoisomerasa I y se demostró que el indotecán tiene un mejor perfil farmacológico [43].
Sin embargo, a pesar del efecto favorable, topotecan condujo a una reducción inespecífica en la expresión de otras áreas genómicas [40]. Además, el deterioro de la actividad de la topoisomerasa reprime la expresión de varios genes largos relacionados con el autismo in vitro [44]. Por tales razones, los efectos «fuera del objetivo» del uso de inhibidores de la topoisomerasa requieren una consideración cuidadosa.
3.2.3. CRISPR/Cas9
CRISPR/Cas9 se ha utilizado con éxito en estudios preclínicos para mutar la región que codifica la transcripción Ube3a-ATS, bloquear su expresión y «de-silenciar» el alelo paterno Ube3a. Los investigadores examinaron una biblioteca de diferentes ARN guía (ARNg), que se dirigen a áreas reguladoras cercanas o dentro del área de codificación Ube3a-ATS del genoma. De ellos, los gRNA que se dirigen a los clústeres de Snord116 y Snord115 dieron como resultado «de-silenciar» el alelo paterno Ube3a de la forma más eficiente cuando se transdujeron a neuronas corticales de ratones que llevan una fusión entre el alelo paterno de Ube3a y un gen codificador de una proteína amarilla fluorescente [20,45]. Se seleccionó un ARNg, Spjw33, que se dirige simultáneamente a 76 áreas dentro de Snord115 para experimentos adicionales (Figura 2c). Spjw33 redujo selectivamente la transcripción de áreas específicas de Ube3a-ATS, en contraste con los controles tratados con topotecan [45].
Cuando a los ratones que carecen del alelo materno Ube3a se les inyectó intracerebroventricularmente un AAV que lleva un Staphylococcus aureus Cas9 y un ARNg que se dirige a una región similar a Spjw33, se produjo un aumento significativo en la expresión paterna de Ube3a en todo el cerebro, incluidas las neuronas corticales, el hipocampo y el médula espinal (pero no el cerebelo). Los efectos persistieron durante 17 meses después de una sola inyección, como lo confirmó el análisis histológico de las neuronas corticales. Los ratones inyectados dos veces (durante el período embrionario y postnatal temprano) mejoraron las características anatómicas y de comportamiento. Este enfoque también dio como resultado un aumento de la expresión bialélica de Ube3a en neuronas primarias derivadas de progenitores neurales humanos transducidas con ARNg que se dirigen al área del grupo Snord115. Los investigadores observaron la integración del vector en el genoma del huésped en las áreas objetivo [45].
Un estudio reciente mostró que la formación de indel mediada por CRISPR/Cas9 en el área genómica entre Snord115 y el Ube3a paterno puede «de-silenciar» el alelo paterno y restaurar el fenotipo motor y conductual en ratones que carecen de Ube3a materno (Figura 2c). A los ratones recién nacidos se les inyectó intracerebroventricularmente un vector AAV que portaba S. aureus Cas9 y el gRNA (ATS-GE) bajo el control del promotor de sinapsina para impulsar la expresión neuronal. El análisis de secuenciación mostró que aproximadamente el 20% de las neuronas se sometieron a edición de genes, lo que sugiere que la edición de genes en un subconjunto de neuronas es adecuada para alterar las características fenotípicas. Los investigadores sugirieron una pausa de la transcripción de Ube3a-ATS en los sitios de inserción indel, lo que permite la transcripción de Ube3a [46]. Las diferencias entre el genoma murino y el humano no permitirán el uso de las mismas secuencias de gRNA para aplicaciones humanas.
Los investigadores también están estudiando el potencial de usar CRISPR/Cas13 para apuntar directamente al ARN de UBE3A-ATS, en lugar de su área de ADN codificante (fuente adicional 12).
3.2.4. Factores de transcripción artificiales
Los factores de transcripción artificial (ATF) son sistemas binarios que consisten en una región de unión al ADN y un efector que puede regular los niveles de expresión del gen objetivo [47]. Los ATF basados en dedos de zinc atraviesan con éxito la barrera hematoencefálica cuando se inyectan por vía subcutánea o intraperitoneal y suprimen la expresión de Ube3a-ATS en modelos de ratón SA [21,48]. La administración sistémica de un ATF compuesto por un dominio de dedo de zinc fusionado con el represor de la transcripción de la caja asociada de Krüppel (KRAB), la proteína TAT que penetra en las células del virus de la inmunodeficiencia humana (VIH) (para facilitar la endocitosis), y una señal nuclear dio como resultado la distribución a través del cerebro, confirmado por fluorescencia e inmunoquímica in vivo. El ATF suprimió la producción de Ube3a-ATS al unirse al área del promotor Snurf / Snrpn (Figura 2d). Esto condujo a la restauración de Ube3a a niveles intermedios entre los ratones SA y los ratones de tipo salvaje, según lo confirmado por la inmunoquímica del hipocampo y el cerebelo, así como por la inmunotransferencia. Diferentes regímenes de dosificación o una combinación de dedos de zinc dirigidos a diferentes áreas promotoras aguas arriba de UBE3A-ATS podrían aumentar la eficacia de este tipo de tratamiento [48]. No se han realizado experimentos de comportamiento para evaluar el efecto sobre los fenotipos de los ratones tratados.
3.2.5. Horquillas cortas de ARN y microARN
Se pueden usar horquillas cortas de ARN (shRNA) y microARN (miARN) para apuntar a la transcripción UBE3A-ATS para su degradación a través del proceso de interferencia del ARN. Se pueden usar vectores virales (por ejemplo, AAV) o plásmidos de ADN para inducir la producción de shRNA en neuronas. La estabilidad de los shRNAs en el entorno celular hace que este tipo de agente sea un candidato prometedor para su uso en tratamientos que no requieran de una frecuencia de dosificación alta. Las terapias para SA que utilizan shRNA (por ejemplo, TSHA-106, OV882) y miRNA están en el nivel de descubrimiento (fuente adicional 13-15).
3.2.6. Pequeñas moléculas
Se demostró que tres compuestos de moléculas pequeñas son eficaces para «de-silenciar» el UBE3A paterno (fuente adicional 16). Estos compuestos parecen tener mejores perfiles de seguridad que los inhibidores de topoisomerasa. Estos compuestos se encuentran en la etapa de descubrimiento de desarrollo. Se requieren más estudios para evaluar la eficacia, biodisponibilidad y perfil de seguridad de estas moléculas.
- Dianas terapéuticas
Los tipos de agentes terapéuticos discutidos anteriormente tienen como objetivo proporcionar un tratamiento definitivo para el SA mediante la restauración de la función UBE3A en las neuronas. Una alternativa es apuntar a rutas moleculares y proteínas efectoras que se conoce que están involucradas en la patofisiología del SA. Los objetivos de estos tratamientos dianas son restaurar la transmisión inhibitoria y mejorar la función sináptica y la plasticidad. Algunos otros tratamientos posteriores se dirigen a vías asociadas con síntomas específicos, como la epilepsia o el mioclono no epiléptico. Los compuestos, empresas o instituciones involucradas y etapas de desarrollo se enumeran en la Tabla 3.
Tabla 3. Dianas terapéuticas
3.3.1. Restauración de la inhibición tónica: gaboxadol (OV101)
Los estudios preclínicos en ratones deficientes en Ube3a materna mostraron que la causa probable de la disfunción motora radica en la alteración funcional de la corteza cerebelosa debido a la inhibición tónica deficiente [49,50]. La electrofisiología de células granulares de cortes cerebelosos reveló que la corriente asociada al receptor de ácido γ-aminobutírico tipo A (GABAA) disminuyó significativamente hasta la edad adulta. En ratones SA, los niveles del transportador de ácido γ-aminobutírico GAT1, que se cree que está ubiquitinado por UBE3A, son altos, y esto da como resultado una excesiva regulación a la baja de los receptores GABAA [49]. La restauración in vivo de los niveles de GABA mediante la administración del compuesto 4,5,6,7-tetrahidroisotiazol- [5,4-c] -piridin-3-ol, un agonista extrasináptico de GABAA resultó en un rescate fenotípico [49].
Se desarrolló un modulador alostérico positivo del receptor delta (δ) -GABA, gaboxadol (OV101), con el objetivo de restaurar la inhibición tónica en pacientes con SA. En un ensayo clínico de fase 2 (STARS, NCT02996305), se descubrió que el gaboxadol era seguro en general y sólo presentaba efectos adversos leves a moderados [51]. Después de 12 semanas de tratamiento, los participantes que fueron tratados por vía oral con 15 mg de gaboxadol por la noche mostraron una mejora general significativa en la escala CGI, específicamente adaptada para SA, en comparación con los controles tratados con placebo. Sin embargo, no hubo una mejora significativa para los participantes tratados con la dosis diaria más alta de 25 mg de gaboxadol administrada en dos dosis de 10 mg y 15 mg. Los investigadores sugirieron que este podría ser el efecto de la tolerancia desarrollada [51].
Se realizó un ensayo clínico de fase 3 (NEPTUNE, NCT04106557) para evaluar la eficacia del gaboxadol oral administrado una vez al día. Este fue un ensayo aleatorizado, doble ciego, controlado con placebo que utilizó una CGI revisada específica de sa, como criterio de valoración principal. Participaron un total de 97 pacientes con sA. En diciembre de 2020, se anunció que no se cumplió el criterio de valoración principal y que no se observaron cambios significativos en las medidas de resultado secundarias (comunicado de prensa, fuente adicional 17).
3.3.2. Agentes para mejorar el crecimiento, el mantenimiento y la función de las sinapsis
3.3.2.1. Factores de crecimiento similares a la insulina
Los factores de crecimiento similares a la insulina IGF-1 e IGF-2 son importantes para el desarrollo, crecimiento y mantenimiento de las sinapsis en el SNC [52,53]. En estudios preclínicos, un análogo de IGF-1 (NNZ-2566) fue ineficaz (fuente adicional 18). Sin embargo, recientemente se demostró que la administración subcutánea de manosa-6-fosfato (M6P) e IGF-2, los ligandos del receptor de IGF-2 (IGF2R), puede mejorar significativamente la disfunción motora, el deterioro cognitivo y la memoria en ratones deficientes en Ube3a materna debido a una mutación nula [21]. Además, los ratones tratados con IGF-2 mostraron una disminución de las convulsiones inducidas acústicamente [54].
3.3.2.2. Glicina-prolina cíclica (NNZ-2591)
La glicina-prolina cíclica (cGP) es un metabolito natural del IGF-1 que regula la biodisponibilidad del IGF-1 [55,56]. NNZ-2591 es un análogo sintético de cGP que tiene una vida media más larga y una biodisponibilidad mejorada [57]. Según las presentaciones en la cumber FAST (fuente adicional 19), el tratamiento de 6 semanas de ratones SA con NNZ-2591 resultó en una mejora significativa en los déficits motores y cognitivos y disminuyó su actividad convulsiva. Los datos del ensayo clínico de fase 1 (NCT04379869) no mostraron problemas de seguridad en voluntarios sanos en Australia (comunicado de prensa, fuente adicional 20). Está previsto realizar un ensayo clínico de fase 2 para determinar la eficacia en pacientes con SA, síndrome de Phelan-McDermid y síndrome de Pitt Hopkins.
3.3.2.3. Inhibidor de la proteína fosfatasa 2A (LB-100)
El activador de fosfotirosil fosfatasa (PTPA), un activador de la proteína fosfatasa 2 (PP2A), es un sustrato de UBE3A y, por lo tanto, algunos pacientes con SA tienen una actividad de PP2A anormalmente alta. La vía de señalización UBE3A-PTPA-PP2A es crucial durante el desarrollo tanto para la morfogénesis de la columna dendrítica como para la función de las sinapsis excitadoras [58]. Tanto la disminución genética del PTPA como la inhibición farmacológica del PP2A restauraron la morfología de la columna dendrítica en modelos de ratón SA. La evaluación de cortes de cerebro de ratones SA, que fueron tratados con el inhibidor de PP2A LB-100, mostró una transmisión sináptica mejorada en la corteza motora primaria en comparación con los ratones no tratados [21, 58]. Además, las inyecciones intraperitoneales de LB-100 en estos ratones produjeron una mejora significativa en la fuerza muscular, la coordinación motora y el aprendizaje después de 14 días. Se descubrió que LB-100 es seguro en un ensayo clínico de fase 1 (NCT01837667) como tratamiento para adultos con tumores sólidos [59]. Esta pequeña molécula se está probando actualmente por su capacidad para cruzar la barrera hematoencefálica en pacientes con tumores cerebrales (NCT03027388). LB-100 se encuentra actualmente en desarrollo preclínico para SA.
3.3.2.4. Fosfato NSI-189
El fosfato NSI-189, una bencilpiperazina-aminopiridina, es un agente neuroprotector que también ha demostrado estimular la neurogénesis tanto in vitro en células madre neurales derivadas del hipocampo humano como in vivo en hipocampo murino [60,61]. En un ensayo clínico de fase 2 para el tratamiento del trastorno de depresión mayor (NCT02695472), NSI-189 tuvo efectos antidepresivos y procognitivos [62,63]. El potencial terapéutico de NSI-189 para SA se ha probado en estudios preclínicos. La electrofisiología de cortes de hipocampo de ratones SA [22], que fueron tratados con NSI-189, mostró una mejora de la LTP inducida por estimulación de ráfagas theta en la región CA1 [22,64]. Además, los tratados durante 16 días demostraron mejores funciones de aprendizaje y memoria, según se evaluó con el condicionamiento del miedo. Dentro de los 5 días de tratamiento con los ratones SA tratados con NSI-189 habían mejorado la función motora y su rendimiento en el tratamiento incluso superó al de los ratones de tipo salvaje en las dosis más altas. Con unos pocos días de tratamiento, los efectos persistieron durante más de 3 semanas, aunque la vida media del compuesto es de aproximadamente 2 horas en ratones. Mecánicamente, se cree que estos cambios están mediados por la vía TrkB-Akt, que se sabe que está involucrada en la plasticidad sináptica. Es probable que los cambios requieran la transcripción de genes, lo que probablemente explica la dependencia del tiempo de los efectos [64].
3.3.3. Tratamiento de los síntomas
3.3.3.1. SAGE-324
SAGE-324 es un modulador alostérico positivo del receptor GABA con una vida media prolongada, que tiene el potencial de mejorar la transmisión GABAérgica interrumpida y tratar los síntomas del SA, como la epilepsia y las mioclonías no epilépticas. Su eficacia se está probando para un amplio espectro de afecciones neurológicas que se presentan con temblor esencial, como la enfermedad de Parkinson (fuente adicional 21). El compuesto se encuentra actualmente en un ensayo clínico de fase 2, y se administra por vía oral a participantes con temblores esencialmente (NCT04305275). Los participantes están siendo evaluados por The Essential Tremor Rating Assessment Scale, un método de calificación validado para el temblor esencial.
3.3.3.2. Ésteres de cetonas
El aumento de cetonas mediante la restricción de carbohidratos a menos de 10 gramos por día se ha utilizado con éxito para la epilepsia intratable, incluidos los pacientes con SA [65]. Un tratamiento de bajo índice glucémico, que se centra más en los índices glucémicos de los carbohidratos consumidos, mostró que la restricción de carbohidratos de bajo índice glucémico a 40-60 gramos por día para los pacientes con SA era beneficiosa para el tratamiento de las convulsiones [66]. Una alternativa sostenible a la dieta cetogénica, con un cumplimiento más esperado, es la suplementación con ésteres de cetonas. En estudios preclínicos, esto ha mejorado significativamente la carga de convulsiones, el fenotipo conductual y la plasticidad sináptica del hipocampo en ratones AS [67]. En un ensayo clínico de fase 2 se evaluó una formulación para la suplementación exógena con el éster de cetona beta-hidroxibutirato [68].
4. Conclusión
Actualmente, al menos 15 enfoques terapéuticos con potencial para tratar el SA se encuentran en etapas de desarrollo preclínico y clínico. Entre ellos, dos ASO y cinco enfoques de tratamiento posteriores se encuentran en desarrollo clínico temprano. Los enfoques de reemplazo de genes y la terapia celular se encuentran actualmente en un desarrollo de las últimas fases de preclínica. Recientemente, un compuesto destinado a restaurar la inhibición tónica no logró alcanzar el criterio de valoración principal en un ensayo clínico de fase 3. Todavía no existe un tratamiento modificador de la enfermedad disponible para el SA. Sin embargo, creemos que en los próximos años, varios candidatos estarán en desarrollo clínico simultáneamente para SA.
5. Opinión de expertos
La cantidad de desarrollos preclínicos y clínicos para el SA es impresionante. Más de cinco tratamientos modificadores de la enfermedad estarán en desarrollo clínico en 2022-2023. En comparación, en 2016 se llevaron a cabo tres ensayos clínicos para la atrofia muscular espinal (AME), que se considera el paradigma de una enfermedad rara con múltiples desarrollos terapéuticos simultáneos [69,70]. Teniendo en cuenta la rareza de la enfermedad, se espera que la disponibilidad de los pacientes se convierta en un obstáculo para ensayos clínicos posteriores o menos prometedores.
El SA es un trastorno monogénico para el que las terapias genéticas, como las ASO o las terapias de reemplazo génico mediadas por virus, tienen el potencial de modificar la enfermedad. El impacto potencial del primer ASO en desarrollo clínico para SA, GTX-102, parece prometedor. Sin embargo, los participantes experimentaron eventos adversos graves con las dosis más altas probadas, que se asociaron con la inflamación de las meninges y las raíces nerviosas en la región de la administración intratecal, pero finalmente se resolvieron (comunicado de prensa, fuente adicional 9). Se ha demostrado que la administración intratecal de ASO es segura en los casos de AME y esclerosis lateral amiotrófica [71-73]. Con respecto a la seguridad, la principal ventaja de los ASO es su alta especificidad por la que se pueden evitar los efectos «fuera del objetivo». En contraste, los inhibidores de la topoisomerasa y los enfoques de ingeniería del genoma podrían ser eficientes para «de-silenciar» la copia paterna, pero son potencialmente menos seguros desde esta perspectiva.
Una terapia génica ex vivo mediada por virus (terapia celular) que utiliza AAV se encuentra actualmente en fases finales de desarrollo preclínico, cerca de las pruebas clínicas. Esta será la primera vez que se pruebe un enfoque terapéutico de este tipo para el SA. Un desafío importante a la hora de traducir los resultados de las terapias génicas mediadas por virus de ratones y primates no humanos a humanos es el escalado de dosis; esto es particularmente desafiante para el SA, ya que se requiere UBE3A en todo el cerebro y el umbral de expresión necesario para el rescate fenotípico sigue siendo desconocido. Las vías de administración más sencillas para tales terapias en el caso de SA son las que se dirigen al espacio subaracnoideo, por vía intratecal por punción lumbar o por inyección intracisterna magna e intracerebroventricular. Ciertamente, se puede lograr una biodisponibilidad mejorada en el SNC usando estas rutas; sin embargo, son altamente intervencionistas, especialmente considerando la probabilidad de que se requiera una nueva dosis. Sin embargo, los AAV tienen una buena capacidad de transducción con neuronas. Además, su genoma suele permanecer como episomas extracromosómicos en las células transducidas y no se incorpora al genoma del huésped [74]. Esto es tranquilizador desde el punto de vista de la seguridad, pero suscita preocupaciones sobre la durabilidad de la expresión. Por el contrario, la próxima terapia génica ex vivo presenta la ventaja de ser un tratamiento permanente: mediante el uso de programación lentiviral, el gen UBE3A se integra en los cromosomas de las MSC y, por lo tanto, se copia con divisiones celulares.
Se esperan desafíos importantes relacionados con la respuesta inmune del huésped, la inflamación y la citotoxicidad subsiguiente, no solo en el desarrollo clínico de los ASO y las terapias de reemplazo génico. Aunque se considera que los AAV tienen un mejor perfil de inmunogenicidad en comparación con otros vectores virales (como los adenovirus), es necesario determinar su seguridad. Hasta ahora, la única terapia de reemplazo génica aprobada para un trastorno neurológico pediátrico es el onasemnogene abeparvovec para la AME. En un estudio en animales de primates y lechones no humanos, utilizando el mismo serotipo AAV y construcción de terapia génica que el onasemnogene abeparvovec, se demostró que su administración conducía a la degeneración de los cuerpos celulares de los ganglios de la raíz dorsal y sus axones [75]. A continuación, la FDA suspendió el ensayo clínico de la forma intratecal de onasemnogene abeparvovec (comunicado de prensa, fuente adicional 22). La adición de dianas de miR183 en los vectores podría ayudar a reducir la expresión transgénica y, por tanto, la toxicidad, en los ganglios de la raíz dorsal [76]. Este enfoque tiene el potencial de lograr una mejor transducción en el cerebro sin el paso limitante de la velocidad de la toxicidad de los ganglios de la raíz dorsal.
Se han realizado estudios preclínicos en modelos animales con mutaciones o deleciones del gen materno. Los pacientes con genotipos distintos a la mutación materna o deleción de UBE3A deben inscribirse cuidadosamente en ensayos clínicos, teniendo en cuenta el mecanismo de acción de la terapia bajo prueba. Por ejemplo, en los casos de disomía uniparental paterna, las estrategias que apuntan a «de-silenciar» la copia paterna podrían teóricamente conducir a la expresión de la proteína UBE3A a niveles tóxicos de ambas copias de genes. La principal ventaja de los tratamientos posteriores es que, en teoría, son activos en todos los genotipos con el mismo paquete de toxicología. Aunque ninguno de estos tratamientos posteriores proporcionará una terapia definitiva para el SA, esperamos que mejoren los síntomas y la calidad de vida en combinación con los enfoques de tratamiento iniciales.
Actualmente, la identificación de medidas de resultado apropiadas con el potencial de servir como criterios de valoración en ensayos clínicos sigue siendo una de las principales necesidades no satisfechas en el SA. Con el fin de prepararse para tales ensayos clínicos, la comunidad de SA estableció el Consorcio de Medición de Resultados y Biomarcadores de SA (ABOM) para garantizar que se avanza con este requisito. Además, se han iniciado estudios de historia natural para recopilar datos de referencia longitudinales de pacientes con SA, así como para crear un entorno de fácil acceso para la ejecución de ensayos clínicos: un estudio dirigido por el Boston Children’s Hospital de la Universidad de Harvard (EE. UU.) Se ha estado ejecutando durante más de 8 años; otro se está iniciando actualmente en la Universidad de Oxford (Reino Unido). Además, como precursor de los ensayos clínicos, se han establecido dos estudios de biomarcadores no intervencionistas. El primero (NCT04103333), que se centra en la identificación de biomarcadores del líquido cefalorraquídeo, está actualmente en curso. El segundo (estudio FREESIAS), que tiene como objetivo identificar medidas de resultado con potencial para convertirse en criterios de valoración en los próximos ensayos clínicos, se centra en una variedad de dominios que incluyen el sueño, las convulsiones, el autocuidado independiente y la comunicación expresiva. Las medidas que se están investigando incluyen la electroencefalografía domiciliaria (EEG) y la monitorización del sueño. El análisis de datos está actualmente en curso para este estudio cerrado y totalmente inscrito.
Los resultados innovadores y específicos del SA se encuentran actualmente en etapas de validación. Un ejemplo es la herramienta ORCA desarrollada por la Universidad de Duke para evaluar el dominio de la comunicación para SA. También se están investigando biomarcadores de EEG, como la frecuencia delta [77]. Además, se demostró que las medidas de movimiento espontáneo, capturadas utilizando tecnología magneto-inercial, eran medidas de resultado muy precisas y sensibles en las poblaciones de distrofia muscular de Duchenne y AME [78,79]. Asimismo, se obtuvieron datos preliminares y muy alentadores para los pacientes con SA (manuscrito en revisión). Estos métodos pueden proporcionar a los investigadores clínicos una plataforma de resultados digitales que podrían usarse en diferentes etapas del desarrollo clínico.
Los desarrollos recientes en AME han demostrado que los fármacos que aportan beneficios menores pero significativos en pacientes postsintomáticos pueden ser transformadores cuando se administran antes de la aparición de los síntomas [80], lo que llevó a la adición de AME a los programas de cribado neonatal en varios países [81]. Se ha propuesto un concepto similar de una «ventana de tiempo» crítica para la intervención para el SA sobre la base de datos de estudios en animales [80, 82, 83], al menos para algunas funciones fisiológicas [37]. Sin embargo, la AME es una afección neurodegenerativa y los datos clínicos recientes informados después del tratamiento con GTX-102 respaldarían la conclusión de que, por el contrario, en el SA existe la posibilidad de cambios significativos en pacientes de distintas edades y, por lo tanto, el concepto de un desarrollo crítico «ventana de tiempo» debe considerarse individualmente (comunicado de prensa, fuente adicional 9). Independientemente, el desarrollo de métodos de cribado neonatal será de crucial importancia, ya que permitirá realizar ensayos en pacientes presintomáticos.
Agradecimientos
Nos gustaría agradecer a la Dra. Allyson C. Berent por su contribución en la revisión de este artículo. Las figuras son originales y fueron creadas con BioRender.com bajo pago de la suscripción.
Aspectos destacados del artículo
El síndrome de Angelman (AS) es un trastorno genético del neurodesarrollo poco común, que es causado por una deficiencia o una función anormal de la ubiquitina proteína ligasa E3A materna, conocida como proteína UBE3A en el sistema nervioso central.
Varios mecanismos moleculares, incluidas deleciones y mutaciones, pueden afectar el gen materno UBE3A en el cromosoma 15 y, posteriormente, la expresión de una proteína normal. La copia paterna del gen se silencia en las neuronas mediante la impronta genómica. Se cree que el ARN largo no codificante, el UBE3A-ATS, dificulta la expresión del gen UBE3A paterno normal.
Entre las estrategias terapéuticas en proceso del SA se encuentran aquellos enfoques que tienen como objetivo restaurar la proteína UBE3A faltante o no funcional en las neuronas mediante terapias de reemplazo de genes o de reemplazo de enzimas. Una terapia de reemplazo génica mediada por virus adenoasociado se encuentra en un desarrollo preclínico tardío, cerca de las pruebas clínicas.
Otra categoría prometedora de enfoques terapéuticos para el SA se dirige a la transcripción UBE3A-ATS con la intención de «de-silenciar» el gen UBE3A paterno. Esta categoría incluye, entre otros, oligonucleótidos antisentido, inhibidores de topoisomerasa y enfoques de ingeniería del genoma. Dos oligonucleótidos antisentido se encuentran actualmente en ensayos clínicos con un tercero a continuación.
Otros enfoques terapéuticos se dirigen a las vías moleculares posteriores, que se sabe que están involucradas en la patofisiología del SA.
Más de 15 enfoques terapéuticos con potencial para tratar el SA se encuentran actualmente en etapas de desarrollo preclínico y clínico. Todavía no existe un tratamiento modificador de la enfermedad disponible para el SA. Sin embargo, creemos que en los próximos años varios candidatos estarán en desarrollo clínico simultáneamente para el SA.
Este recuadro resume los puntos clave contenidos en el artículo.
Declaración de interese
Los autores no tienen afiliaciones relevantes o participación financiera con ninguna organización o entidad con un interés financiero o conflicto financiero con el tema o materiales discutidos en el manuscrito. Esto incluye empleo, consultorías, honorarios, propiedad u opciones de acciones, testimonio de expertos, donaciones o patentes recibidas o pendientes, o regalías.
Declaraciones del revisor
Los revisores por pares de este manuscrito no tienen ninguna relación financiera o de otro tipo relevante que declarar.
Fuentes adicionales
1.https://www.youtube.com/watch?v=EdZGTeoCILY (Gene Therapy – presentation at FAST Summit 2018)
2.https://www.youtube.com/watch?v=RL1lxU1VfU8 (Gene Therapy – presentation at FAST Summit 2018)
3.https://www.youtube.com/watch?v=cSVxoe2XhaQ (Gene Therapy – presentation at FAST Summit 2019)
4.https://www.youtube.com/watch?v=ePkGrb8UeTA (Gene Therapy – presentation at FAST Summit 2020)
5.https://www.youtube.com/watch?v=3qbqRfrlUdE (Gene Therapy – presentation at FAST Summit 2020)
6.https://www.youtube.com/watch?v=OxEVjlG4oGQ (Cell Therapy – presentation at FAST Summit 2019)
7.https://www.youtube.com/watch?v=xy5jnaL-cYY (Cell Therapy – presentation at FAST Summit 2020)
8.https://cureangelman.org/pilot-feasibility-of-an-enzyme-replacement-therapy-for-as (ERT)
9.https://ir.ultragenyx.com/news-releases/news-release-details/genetx-and-ultragenyx-announce-presentation-phase-12-data (press release for GTX-102)
10.https://forpatients.roche.com/en/trials/neurodevelopmental-disorder/angelman-syndrome/a-study-to-investigate-the-safety–tolerability–pharma-19556.html (RO7248824 or RG6091)
12.https://cureangelman.org/the-difference-between-cas13-and-cas9-in-angelman-syndrome-research (CRISPR/Cas13)
13.https://tayshagtx.com/pipeline/ (shRNA)
14.https://www.angelman.org/for-parents/angelman-therapies/ (shRNA)
16.https://www.angelman.org/research/pilot-study-to-validate-three-novel-classes-of-small-molecules-to-unsilence-paternal-UBE3A-allele/ (small molecules)
17.https://www.globenewswire.com/news-release/2020/12/01/2137913/0/en/Ovid-Therapeutics-Announces-Phase-3-NEPTUNE-Clinical-Trial-of-OV101-for-the-Treatment-of-Angelman-Syndrome-Did-Not-Meet-Primary-Endpoint.html (press release for OV101/gaboxadol)
18.https://cureangelman.org/toward-therapeutics-the-fire-team-series-part-iv (IGF-1 analog)
19.https://www.youtube.com/watch?v=_AV-SKR6nJw (NNZ-2591 – presentation at FAST Summit 2019)
20.https://www.neurenpharma.com/irm/PDF/29bba6b7-7f35-4107-9c8b-27833803e8dc/SuccessfulPhase1trialforNeuren39sNNZ2591 (press release for NNZ-2591)
22.https://www.novartis.com/news/media-releases/novartis-announces-avxs-101-intrathecal-study-update (press release for onasemnogene abeparvove)
Información Adicional
Financiación
Este trabajo fue apoyado por FAST UK y la fundación Onassis, de la cual T. Markati es docente (Identificación de la beca: F ZQ 040-1 / 2020-2021).
Referencias
Los artículos de especial interés se han destacado como de interés (•) o de considerable interés (••) para los lectores.
- Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385–395. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Thibert RL, Larson AM, Hsieh DT, et al. Neurologic manifestations of Angelman syndrome. Pediatr Neurol. 2013;48(4):271–279. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Larson AM, Shinnick JE, Shaaya EA, et al. Angelman syndrome in adulthood. American Journal of Medical Genetics Part A. 2015;167(2):331–344. [Crossref], [Web of Science ®], [Google Scholar]
- Bird LM. Angelman syndrome: review of clinical and molecular aspects. The Application of Clinical Genetics. 2014;93. DOI:10.2147/TACG.S57386 [Crossref], [PubMed], [Google Scholar]
- Dagli A, Buiting K, Williams CA. Molecular and clinical aspects of Angelman syndrome. Mol Syndromol. 2012;2(3–5):100–112. [PubMed], [Google Scholar]
- Kyllerman M. Angelman syndrome. In: Handbook of clinical neurology. 2013;111:287-290. [Google Scholar]
- Sadhwani A, Wheeler A, Gwaltney A, et al. Developmental skills of individuals with angelman syndrome assessed using the bayley-III. J Autism Dev Disord. 2021. 10.1007/s10803-020-04861-1. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Wheeler AC, Sacco P, Cabo R. Unmet clinical needs and burden in Angelman syndrome: a review of the literature. Orphanet J Rare Dis. 2017;12(1). 10.1186/s13023-017-0716-z [Crossref], [PubMed], [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15(1):70–73. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Matsuura T, Sutcliffe JS, Fang P, et al. De novo truncating mutations in E6-Ap ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15(1):74–77. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Buiting K, Williams C, Horsthemke B. Angelman syndrome — insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12(10):584–593. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Meng L, Person RE, Beaudet AL. Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet. 2012;21(13):3001–3012. [Crossref], [Web of Science ®], [Google Scholar]
• UBE3A-ATS media el «silenciamiento» de la copia paterna UBE3A
- Meng L, Person RE, Huang W, et al., Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the angelman syndrome mouse model. PLoS Genet. 2013; 9(12): e1004039. [Crossref], [Web of Science ®], [Google Scholar]
• La focalización del UBE3A-ATS tiene el potencial de «de-silenciar» la copia paterna del UBE3A.
- Lee SY, Ramirez J, Franco M, et al. Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell Mol Life Sci. 2014;71(14):2747–2758. [Crossref], [Web of Science ®], [Google Scholar]
- Jiang YH, Beaudet AL. Human disorders of ubiquitination and proteasomal degradation. Curr Opin Pediatr. 2004;16(4):419–426. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Yang X. Towards an understanding of Angelman syndrome in mice studies. J Neurosci Res. 2020;98(6):1162–1173. [Crossref], [Web of Science ®], [Google Scholar]
- Sun J, Liu Y, Zhu G, et al. PKA and Ube3a regulate SK2 channel trafficking to promote synaptic plasticity in hippocampus: implications for angelman syndrome. Sci Rep. 2020;10:9824. [Web of Science ®], [Google Scholar]
- Martínez-Noël G, Luck K, Kühnle S, et al. Network Analysis of UBE3A/E6AP-associated proteins provides connections to several distinct cellular processes. J Mol Biol. 2018;430(7):1024–1050. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Yashiro K, Riday TT, Condon KH, et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. 2009;12(6):777–783. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Dindot SV, Antalffy BA, Bhattacharjee MB, et al. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17(1):111–118. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21(4):799–811. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Baudry M, Kramar E, Xu X, et al. Ampakines promote spine actin polymerization, long-term potentiation, and learning in a mouse model of Angelman syndrome. Neurobiol Dis. 2012;47(2):210–215. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Cheron G, Servais L, Wagstaff J, et al. Fast cerebellar oscillation associated with ataxia in a mouse model of angelman syndrome. Neuroscience. 2005;130(3):631–637. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Daily JL, Nash K, Jinwal U, et al., Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS ONE. 2011; 6(12):e27221. [Crossref], [Web of Science ®], [Google Scholar]
•• Prueba de concepto para la terapia de reemplazo génico.
- Cheron G, Servais L, Dan B. Cerebellar network plasticity: from genes to fast oscillation. Neuroscience. 2008;153(1):1–19. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. 2020;21(4):255–272. [Crossref], [Web of Science ®], [Google Scholar]
- Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–378. [Crossref], [Web of Science ®], [Google Scholar]
- Sirois CL, Bloom JE, Fink JJ, et al. Abundance and localization of human UBE3A protein isoforms. Hum Mol Genet. 2020;48(4):271–279. [Google Scholar]
- Adhikari A, Copping NA, Beegle J, et al. Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum Mol Genet. Internet]. 2021 [cited 2021 Apr 22]; Available from:https://pubmed.ncbi.nlm.nih.gov/33856035/. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
•• Prueba de concepto para terapia celular.
- Garcia-Perez L, Ordas A, Canté-Barrett K, et al. Preclinical development of autologous hematopoietic stem cell-based gene therapy for immune deficiencies: a journey from mouse cage to bed side. Pharmaceutics. 2020;12(6):549. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. [Crossref], [Web of Science ®], [Google Scholar]
- Shemesh E, Deroma L, Bembi B, et al. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst Rev. 2015. DOI:10.1002/14651858.CD010324.pub2. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Angelini C, Semplicini C. Enzyme replacement therapy for pompe disease. Curr Neurol Neurosci Rep. 2012;12(1):70–75. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Dodge A, Willman J, Willman M, et al. Identification of UBE3A Protein in CSF and extracellular space of the hippocampus suggest a potential novel function in synaptic plasticity. Autism Res. 2021;14(4):645–655. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Galiveti CR, Raabe CA, Konthur Z, et al. Differential regulation of non-protein coding RNAs from prader-willi syndrome locus. Sci Rep. 2014;4. 10.1038/srep06445. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Chamberlain SJ, Brannan CI. The Prader–willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics. 2001;73(3):316–322. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Meng L, Ward AJ, Chun S, et al., Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518(7539): 409–412. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
•• Prueba de concepto para el uso de oligonucleótidos antisentido para «de-silenciar» la copia paterna de UBE3A
- Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14(1):9–21. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Gr??nweller A, Hartmann RK. Locked nucleic acid oligonucleotides: the next generation of antisense agents? BioDrugs. 2007;21(4):235–243. [Crossref], [Web of Science ®], [Google Scholar]
- Huang HS, Allen JA, Mabb AM, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481(7380):185–189. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Powell WT, Coulson RL, Gonzales ML, et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(34):13938-13943. [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ. A double-edged sword: r loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014;28(13):1384–1396. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Lee HM, Clark EP, Kuijer MB, et al. Characterization and structure-activity relationships of indenoisoquinoline-derived topoisomerase i inhibitors in unsilencing the dormant Ube3a gene associated with Angelman syndrome. Mol Autism. 2018;9(1). 10.1186/s13229-018-0228-2. [Crossref], [Google Scholar]
- King IF, Yandava CN, Mabb AM, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501(7465):58–62. [Crossref], [Web of Science ®], [Google Scholar]
- Wolter JM, Mao H, Fragola G, et al., Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature.2020; 587(7833): 281–284. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
•• Prueba de concepto de CRISPR / Cas9 para “de-silenciar” la copia paterna de UBE3A
- Schmid RS, Deng X, Panikker P, et al. CRISPR/Cas9 directed to the Ube3a antisense transcript improves Angelman syndrome phenotype in mice. J Clin Investig. 2021; 131(5). DOI: 10.1172/JCI142574 [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
•• Prueba de concepto de CRISPR / Cas9 para “de-silenciar” la copia paterna de UBE3A
- Sera T. Zinc-finger-based artificial transcription factors and their applications. Adv Drug Deliv Rev. 2009;61(7–8):513–526. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Bailus BJ, Pyles B, Mcalister MM, et al. Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an angelman syndrome mouse brain. Mol Ther. 2016;24(3):548–555. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Egawa K, Kitagawa K, Inoue K, et al. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of angelman syndrome. Sci Transl Med. 2012;4(163):163ra157–163ra157. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Miura K, Kishino T, Li E, et al. Neurobehavioral and electroencephalographic abnormalities in Ube3aMaternal-deficient mice. Neurobiol Dis. 2002;9(2):149–159. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Bird LM, Ochoa-Lubinoff C, Tan W-H, et al. The STARS Phase 2 Study: a randomized controlled trial of gaboxadol in angelman syndrome. Neurology 2020 10.1212/WNL.0000000000011409 10.1212/WNL.0000000000011409 [Crossref], [Google Scholar]
- Werner H, LeRoith D. Insulin and insulin-like growth factor receptors in the brain: physiological and pathological aspects. Eur Neuropsychopharmacol. 2014;24(12):1947–1953. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- O’Kusky J, Ye P. Neurodevelopmental effects of insulin-like growth factor signaling. Front Neuroendocrinol. 2012;33(3):230-251. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Cruz E, Descalzi G, Steinmetz A, et al. CIM6P/IGF-2 receptor ligands reverse deficits in angelman syndrome model mice. Autism Res. 2021;14(1):29–45. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Guan J, Gluckman P, Yang P, et al. Cyclic glycine-proline regulates IGF-1 homeostasis by altering the binding of IGFBP-3 to IGF-1. Sci Rep. 2014;4:4388. [Web of Science ®], [Google Scholar]
- Guan J, Singh-Mallah G, Liu K, et al. The role for cyclic glycine-proline, a biological regulator of insulin-like growth factor-1 in pregnancy-related obesity and weight changes. J Biol Regul Homeost Agents. 2018;32(3):465–478. [Web of Science ®], [Google Scholar]
- Guan J, Zhang R, Dale-Gandar L, et al. NNZ-2591, a novel diketopiperazine, prevented scopolamine-induced acute memory impairment in the adult rat. Behav Brain Res. 2010;210(2):221–228. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Wang J, Sen LS, Wang T, et al. UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology. Proceedings of the National Academy of Sciences of the United States of America. 2019;116 (25):12500-12505 [Crossref], [Google Scholar]
- Chung V, Mansfield AS, Braiteh F, et al. Safety, tolerability, and preliminary activity of LB-100, an inhibitor of protein phosphatase 2A, in patients with relapsed solid tumors: an open-label, dose escalation, first-in-human, phase I trial. Clin Cancer Res. 2017;23(13):3277–3284. [Crossref], [Web of Science ®], [Google Scholar]
- Allen BD, Acharya MM, Lu C, et al. Remediation of radiation-induced cognitive dysfunction through oral administration of the neuroprotective compound NSI-189. Radiat Res. 2018;189(4):345. [Crossref], [Web of Science ®], [Google Scholar]
- McIntyre RS, Johe K, Rong C, et al. The neurogenic compound, NSI-189 phosphate: a novel multi-domain treatment capable of pro-cognitive and antidepressant effects. Expert Opin Investig Drugs. 2017;26(6):767–770. [Taylor & Francis Online], [Web of Science ®], [Google Scholar]
- Fava M, Johe K, Ereshefsky L, et al. A Phase 1B, randomized, double blind, placebo controlled, multiple-dose escalation study of NSI-189 phosphate, a neurogenic compound, in depressed patients. Mol Psychiatry. 2016;21(10):1483-1484. [Web of Science ®], [Google Scholar]
- Papakostas GI, Johe K, Hand H, et al. A phase 2, double-blind, placebo-controlled study of NSI-189 phosphate, a neurogenic compound, among outpatients with major depressive disorder. Mol Psychiatry. 2020;25(7):1569–1579. [Crossref], [Web of Science ®], [Google Scholar]
- Liu Y, Johe K, Sun J, et al. Enhancement of synaptic plasticity and reversal of impairments in motor and cognitive functions in a mouse model of Angelman Syndrome by a small neurogenic molecule, NSI-189. Neuropharmacology. 2019;144:337–344. [Crossref], [Web of Science ®], [Google Scholar]
- Evangeliou A, Doulioglou V, Haidopoulou K, et al. Ketogenic diet in a patient with Angelman syndrome. Pediatr Int. 2010;52(5):831–834. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Thibert RL, Pfeifer HH, Larson AM, et al. Low glycemic index treatment for seizures in Angelman syndrome. Epilepsia. 2012;53(9):1498–1502. [Crossref], [Web of Science ®], [Google Scholar]
- Ciarlone SL, Grieco JC, D’Agostino DP, et al. Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol Dis. 2016;96:38–46. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Herber DL, Weeber EJ, D’Agostino DP, et al. Evaluation of the safety and tolerability of a nutritional Formulation in patients with ANgelman Syndrome (FANS): study protocol for a randomized controlled trial. Trials. 2020;21(1). DOI:10.1186/s13063-019-3996-x. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Ramdas S, Servais L. New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother. 2020;21(3):307–315. [Taylor & Francis Online], [Web of Science ®], [Google Scholar]
- Servais L, Baranello G, Scoto M, et al. Therapeutic interventions for spinal muscular atrophy: preclinical and early clinical development opportunities. Expert Opin Investig Drugs. 2021; Internet]. [cited 2021 Apr 22];1–9. Available from: https://pubmed.ncbi.nlm.nih.gov/33749510/. [PubMed], [Web of Science ®], [Google Scholar]
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–1732. [Crossref], [Web of Science ®], [Google Scholar]
- Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–635. [Crossref], [Web of Science ®], [Google Scholar]
- Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109–119. [Crossref], [Web of Science ®], [Google Scholar]
- Deyle DR, Russell DW. Adeno-associated virus vector integration. Curr Opin Mol Ther. 2009;11(4):442–447. [PubMed], [Google Scholar]
- Hinderer C, Katz N, Buza EL, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29(3):285–298. [Crossref], [Web of Science ®], [Google Scholar]
- Hordeaux J, Buza EL, Jeffrey B, et al. MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates. Sci Transl Med. 2020;12(569):eaba9188. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Sidorov MS, Deck GM, Dolatshahi M, et al. Delta rhythmicity is a reliable EEG biomarker in Angelman syndrome: a parallel mouse and human analysis. J Neurodev Disord. 2017;9:article number:17. [Web of Science ®], [Google Scholar]
- Lilien C, Gasnier E, Gidaro T, et al. Home-based monitor for gait and activity analysis. J Visualized Exp. 2019(150). doi:10.3791/59668. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Annoussamy M, Seferian AM, Daron A, et al. Natural history of Type 2 and 3 spinal muscular atrophy: 2‐year NatHis‐SMA study. Ann Clin Transl Neurol. 2020;8(2):359–373. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Dangouloff T, Servais L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag. 2019;15:1153–1161. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Dangouloff T, Burghes A, Tizzano EF, et al. 244th ENMC international workshop: newborn screening in spinal muscular atrophy May 10–12, 2019, Hoofdorp, The Netherlands. Neuromuscul Disord. 2020;30(1):93–103. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Silva-Santos S, Van Woerden GM, Bruinsma CF, et al. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Investig. 2015;125(5):2069–2076. [Crossref], [PubMed], [Web of Science ®], [Google Scholar]
- Sonzogni M, Hakonen J, Bernabé Kleijn M, et al. Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol Autism. 2019;10(1). DOI:10.1186/s13229-019-0277-1. [Crossref], [PubMed], [Google Scholar]